
Historia y Descubrimiento

El Síndrome de Apert, también conocido como acrocefalosindactilia, fue descrito por primera vez en 1906 por el médico francés Eugène Apert. En ese momento, Apert identificó a nueve pacientes con características comunes, incluyendo una cabeza puntiaguda (acrocefalia) y la fusión de dedos en manos y pies (sindactilia). El término “acrocefalosindactilia” proviene de las palabras griegas “acro” para “cumbre” (refiriéndose a la cabeza puntiaguda) y “sindactilia” para “fusión de dedos”. En honor a su descubridor, la condición se conoce como Síndrome de Apert1.
Definición y Causas
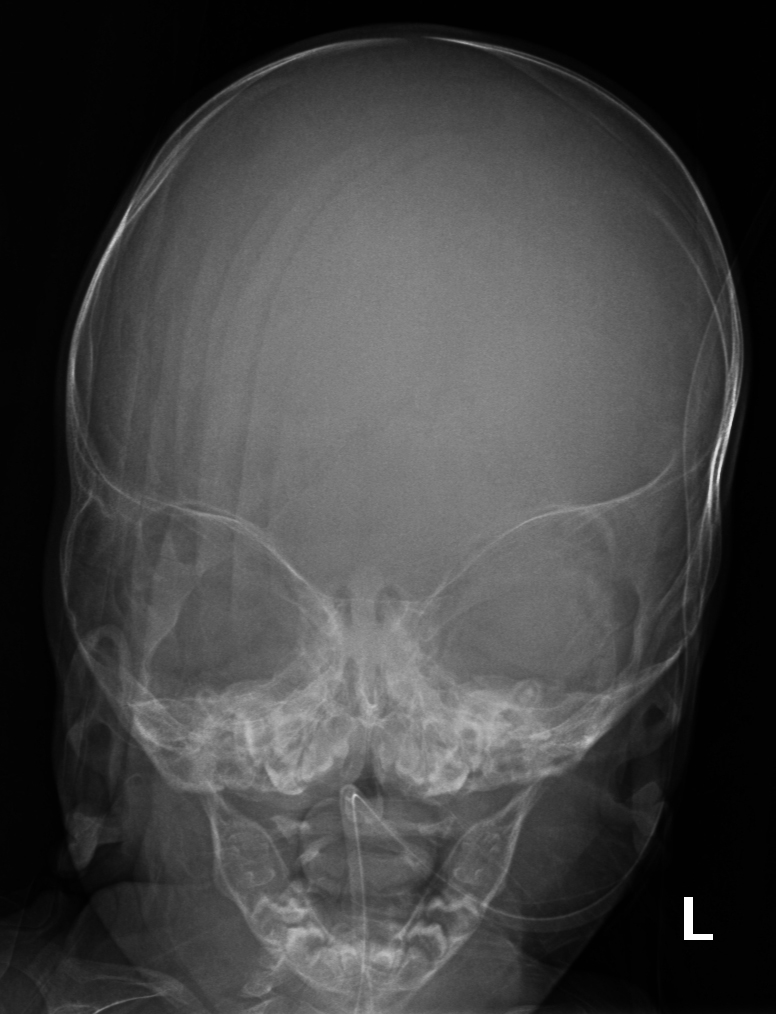
El Síndrome de Apert es un trastorno genético autosómico dominante causado por una mutación espontánea en el gen FGFR2, específicamente en el receptor 2 del factor de crecimiento de los fibroblastos. Esta anomalía genética provoca el cierre prematuro de las suturas entre los huesos del cráneo, una condición conocida como craneosinostosis, afectando la forma de la cabeza y la cara2.
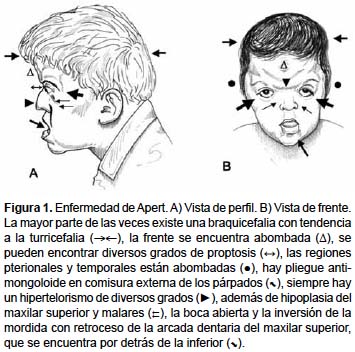
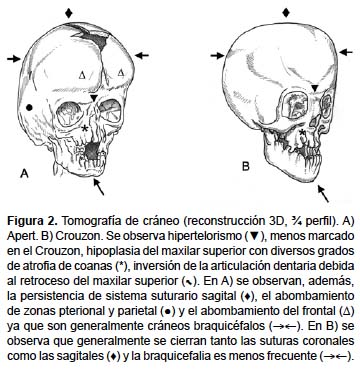
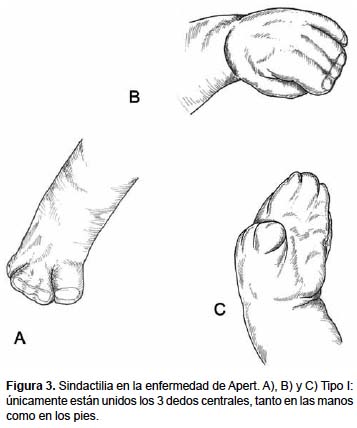
Tasa de Incidencia y Estadísticas en México
Según las estadísticas de la Secretaría de Salud de México, el Síndrome de Apert afecta a aproximadamente 1 de cada 160,000 recién nacidos en el país. Esta cifra proporciona una visión más específica de la prevalencia de la condición en la población mexicana y destaca la importancia de la concientización y el apoyo².
Síntomas y Manifestaciones Clínicas
Los síntomas del Síndrome de Apert pueden variar, pero comúnmente incluyen:
- Craneosinostosis: Cierre prematuro de las suturas craneales, evidenciado por crestas a lo largo de las suturas.
- Infecciones del oído: Problemas recurrentes en el oído, que requieren atención médica continua.
- Deformidades en las manos y pies: Incluyendo sindactilia, donde los dedos están fusionados.
- Pérdida de la audición: Un efecto secundario que puede requerir intervención especializada.
- Fontanela grande: Punto blando en el cráneo del bebé que puede cerrarse más tarde de lo esperado.
- Desarrollo intelectual variado: Puede haber retraso en el desarrollo cognitivo.
- Características faciales distintivas: Como ojos prominentes, puente nasal plano y labios finos.
- Altura baja y otras anomalías esqueléticas.



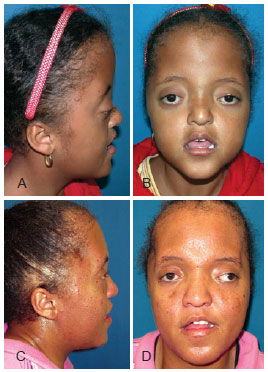
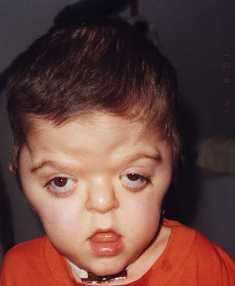
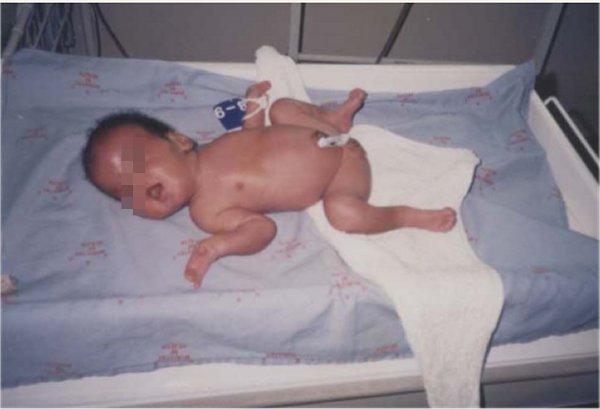
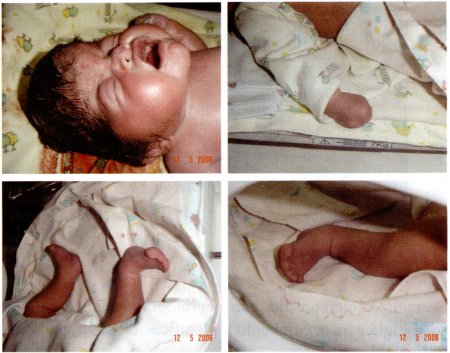
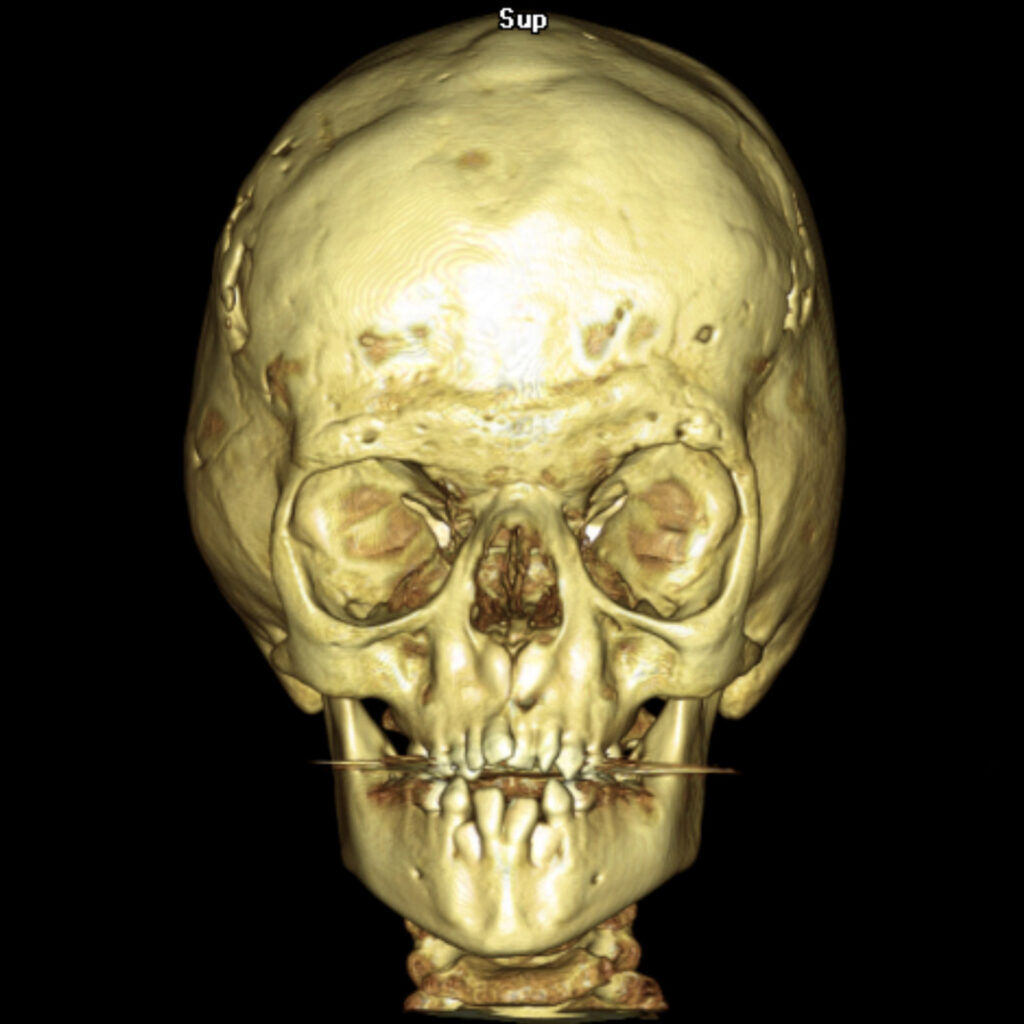
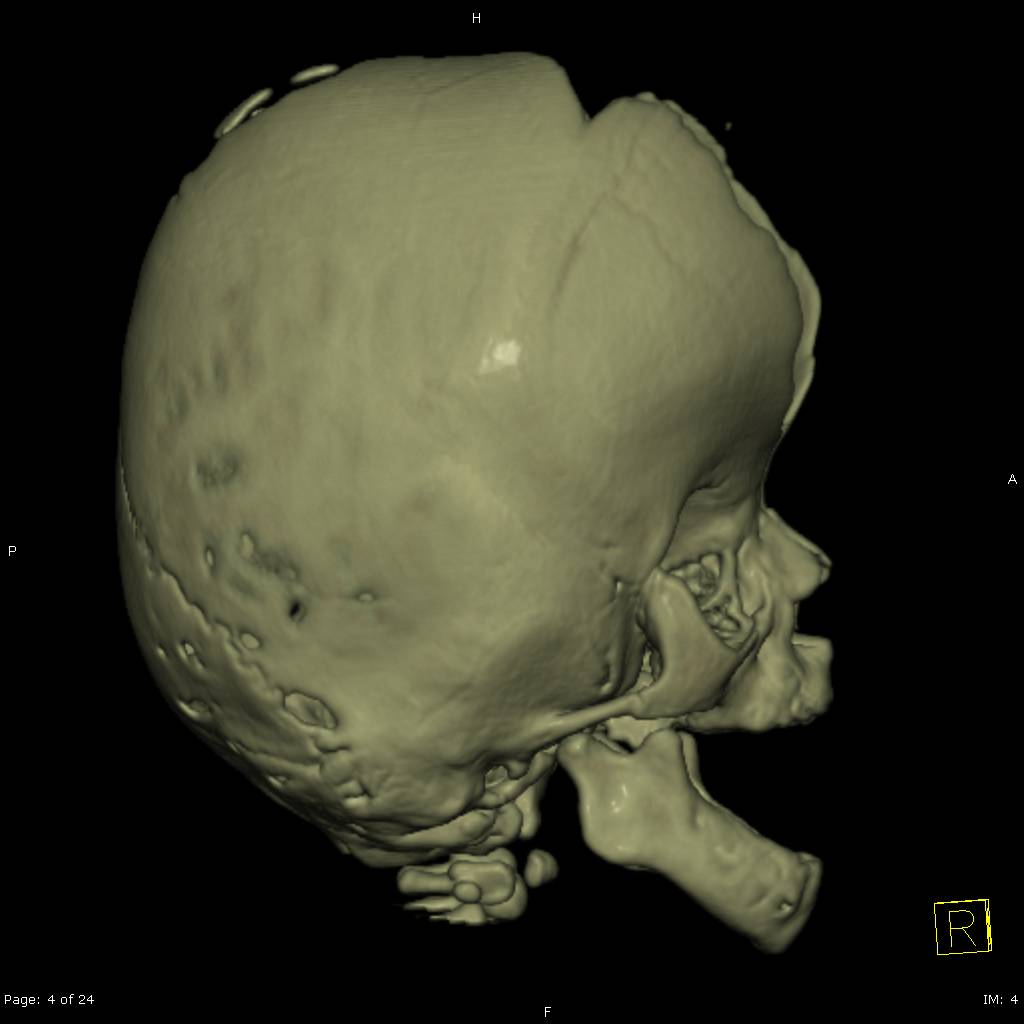
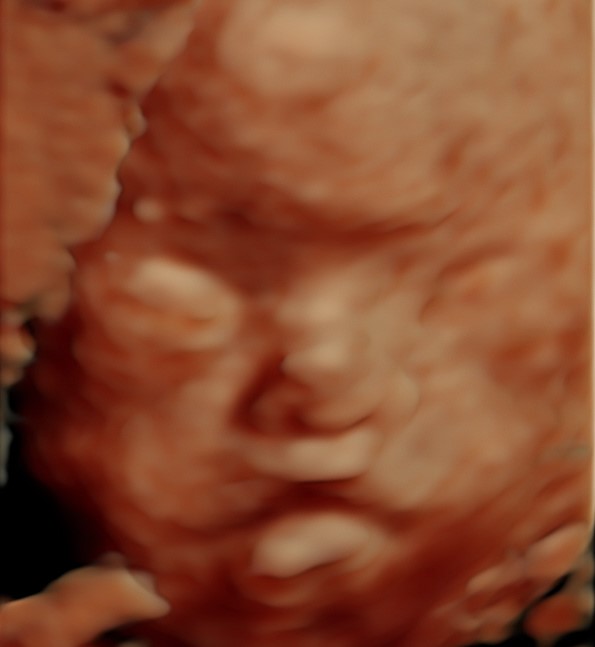
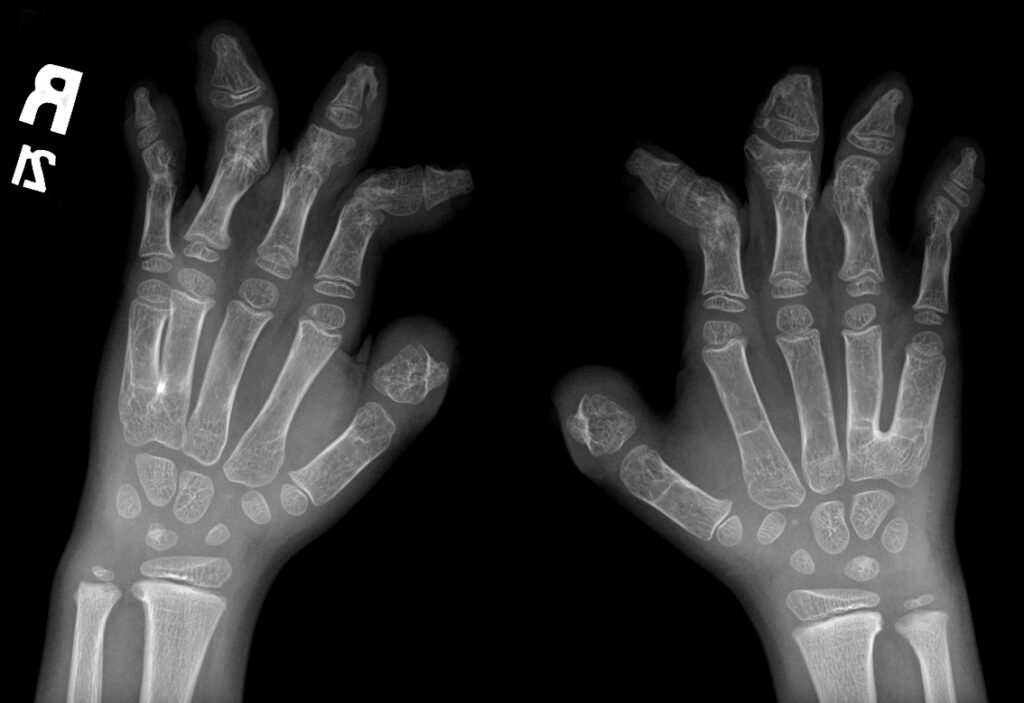
Diagnóstico y Tratamiento
El diagnóstico del Síndrome de Apert se basa en un examen físico detallado, radiografías y pruebas genéticas para identificar la mutación en el gen FGFR2. El tratamiento implica cirugía para corregir craneosinostosis y deformidades en manos y pies. Es crucial la intervención de equipos médicos multidisciplinarios especializados en cirugía craneofacial y genética.
Investigación y Estudios Genéticos
El defecto genético en el Síndrome de Apert afecta al receptor 2 del factor de crecimiento de los fibroblastos (FGFR2). Se calcula una prevalencia de 15 por 1 millón de nacidos vivos, aunque esta cifra es mayor en algunos países asiáticos. La investigación genética y los estudios clínicos son fundamentales para comprender mejor esta condición y desarrollar tratamientos más efectivos.
Manejo y Rehabilitación
El tratamiento rehabilitador incluye programas personalizados de estimulación temprana, terapia ocupacional y técnicas psicopedagógicas. Este enfoque multidisciplinario ha demostrado ser esencial para abordar las áreas del desarrollo cognitivo, social y motor, mejorando así la calidad de vida de los afectados por el Síndrome de Apert.
Diagnóstico Diferencial
El diagnóstico diferencial del Síndrome de Apert implica considerar otras formas de acrocefalosindactilia y acrocefalopolisindactilia. Es crucial distinguir entre estas condiciones para garantizar un tratamiento y manejo adecuados. Algunos de los síndromes relacionados incluyen:
- Síndrome de Saethre-Chotzen: también se caracteriza por craneosinostosis, deformidades en manos y pies, y puede incluir características faciales distintivas. Sin embargo, difiere en la forma en que se manifiestan las fusiones digitales y otras características clínicas específicas.
- Síndrome de Pfeiffer: comparte similitudes con el Síndrome de Apert, como la craneosinostosis y características faciales distintivas. Las fusiones digitales pueden estar presentes, pero el patrón es diferente y puede haber anomalías adicionales, como pulgares anchos y ganchudos.
- Síndrome de Carpenter: se caracteriza por craneosinostosis, manos en forma de zanahoria y pies en forma de raqueta. Aunque comparte algunas características con el Síndrome de Apert, las fusiones digitales y otras malformaciones pueden diferir.
- Síndrome de Crouzon: también involucra craneosinostosis y características faciales distintivas. Sin embargo, se diferencia en la ausencia de sindactilia y en la presentación de otras anomalías craneofaciales específicas.
Es fundamental realizar evaluaciones clínicas y pruebas genéticas detalladas para diferenciar con precisión entre estos síndromes y garantizar un diagnóstico correcto. La colaboración con especialistas en genética y cirugía craneofacial es esencial para un enfoque integral en la identificación y manejo de estas condiciones.
Conclusiones y Reflexiones
El Síndrome de Apert presenta desafíos clínicos únicos, pero con un diagnóstico temprano, intervenciones especializadas y un enfoque integral, se pueden lograr resultados positivos. La colaboración entre profesionales médicos, genetistas y terapeutas es esencial para ofrecer el mejor cuidado a aquellos afectados por esta condición genética. La investigación continua y la concientización son clave para avanzar en la comprensión y el tratamiento del Síndrome de Apert.
Al igual que cada persona, los pacientes con Síndrome Apert, se enfrentan a grandes retos a lo largo de su vida, pero también sueñan, cumplen ideales e inspiran a vivir una lucha constante por la inclusión, igualdad de oportunidades y amor por la vida.
- Hoyos Serrano Maddelainne, Rojas Mamani Jimmy. SINDROME DE APERT (SA). Rev. Act. Clin. Med
- Síndrome de Apert: MedlinePlus enciclopedia médica. ; ¿Qué es el Síndrome de Apert? | Nicklaus Children’s Hospital. ; Síndrome de Apert – Wikipedia, la enciclopedia libre.
- Elsa Camargo Luaces, Zulema Serrano Figueroa. Síndrome de Apert: Revista de Ciencias Médicas de Pinar del Río.